Cart (0 Items)
Your cart is currently empty.
View Products Peptide synthesis
Peptide synthesis
 Philippe Funfrock
Philippe Funfrock
Peptides play a fundamental role as signaling molecules in the regulation of human metabolism. For decades, scientists have understood the potential of using these molecules as therapeutic agents.
The realization of this potential started with the isolation of the first peptides from exogenous sources. The isolation of the first therapeutic peptide was achieved as early as the 1920s by researchers from the University of Toronto. These researchers were able to use purified pancreatic extracts containing insulin for the treatment of diabetes. This exogenous insulin could easily mimic the endogenous molecule and, thus could be applied as “replacement therapy” when insulin’s endogenous concentrations were insufficient.
Insulin, alongside adrenocorticotrophic hormone (ACTH) later purified from pituitary glands, were considered one of the most important life-saving therapeutics during the first part of the 20th century. But it was when the chemical synthesis of peptides became feasible in the 1950s, that researchers and industries finally grasped the true potential of these biopharmaceuticals.
As of 2017, over 60 peptides have been approved for clinics in the United States, Europe, and Japan. And, at this point, more than 150 peptides were in active clinical trials, with an average of 10 to 20 new peptides entering this phase every year since 2010.
Alongside protein-based therapeutic agents, peptides comprise around 10% of the global pharmaceutical market. But this renewed interest in therapeutic peptides was only possible due to the development and subsequent improvement of the chemical synthesis of peptides.
Interestingly, the first fully chemical synthesis of a peptide was reported as early as 1901 by Emil Fischer and Ernest Fourneau. These researchers were able to assemble the first synthetic peptide in the lab from amino acid precursors in a step-wise manner. But it was only after the discovery of the reversible Nα-protecting group, and the development of solid-phase peptide synthesis techniques, that these chemical techniques started to attract more attention from the scientific community.
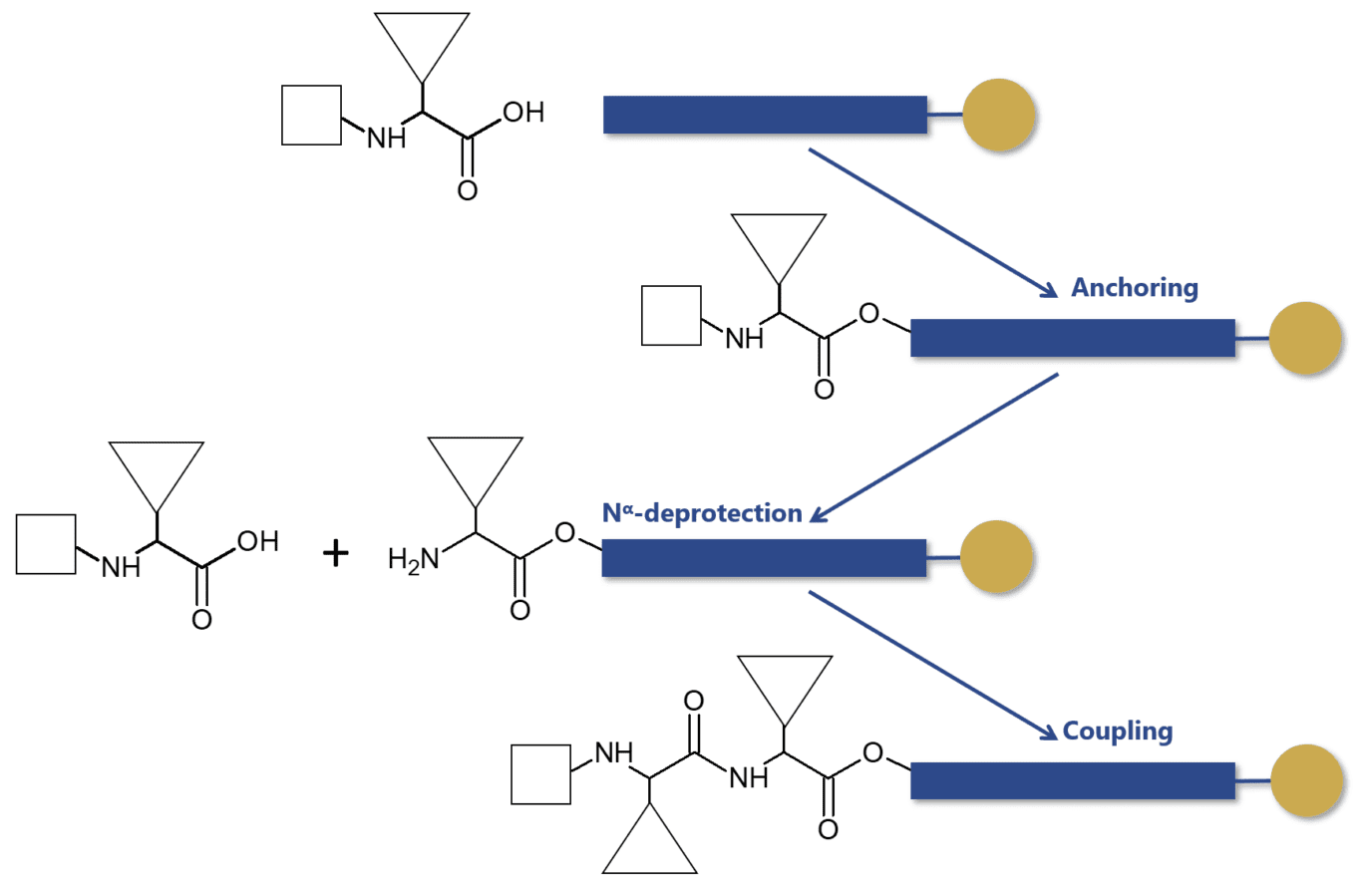
To this day, Bruce Merrifield is still considered the father of modern peptide synthesis. It was Merrifield who revolutionized this process by developing a solid-phase technique in the 1960s. Before his discovery, peptide synthesis was considered a time-consuming and complex process with severe limitations due to the intrinsic instability of peptides in solution. Merrifield changed this by anchoring the peptide chains to an inert and insoluble polymer.
This innovation allowed the full automatization of the entire process and accomplished, for the first time, the chemical synthesis of very large peptides (ribonuclease A with 124 amino acid residues). An accomplishment that was virtually impossible to reproduce using conventional techniques.
His discovery earned him a Nobel Prize in Chemistry in 1984 and has forever changed the potential use of peptides for many different applications.
Even before the discovery of the solid-phase peptide synthesis by Merrifield, researchers were aware that peptides could be assembled in a stepwise manner from the C to N terminus using Nα-protected amino acids. These groups were essential to minimize the side reactions that could reduce the intended size of the peptide and cause branching of the peptide chain.
These groups aimed at conferring temporary protection to the most reactive sites of the peptide building blocks. Thus, they were designed to be easily removed during peptide synthesis.
The first reversible Nα-protecting group for peptide synthesis was described by chemists Max Bergman and Leonidas Zervas as early as 1932. Currently, two of the most relevant N-terminal protecting groups are acid-labile tert-butyloxycarbonyl (Boc) or base-labile 9-fluorenylmethoxycarbonyl (Fmoc). The Boc approach requires the use of strong acids such as trifluoroacetic acid (TFA). while the Fmoc approach requires only the use of mild bases such as piperidine.
Since the Boc chemistry was fine-tuned by Merrifield and his colleagues from the 1960s to the 1980s, the process became a mature technology suited for the synthesis of complex peptides. But due to the harsh acidic conditions required by this chemistry, its use became more restricted after the description of base-labile Fmoc Nα-protecting groups by Louis Carpino and Grace Han in 1970.
Fmoc chemistry also presented a unique set of challenges. However, the replacement of strong acids by mild bases allowed to increase the yield of the synthesis of peptides rich in tryptophan (Trp). These residues were known to suffer extensive modifications in the indole ring due to acid-catalyzed side reactions. These issues lead to very poor synthesis yields (as low as 5%) for Trp-rich peptides such as gramicidin A, an antibiotic peptide. Fmoc chemistry considerably alleviated these issues and increased the yield of gramicidin A up to 87%.
For these reasons, Boc chemistry has been subsequently replaced by the Fmoc approach for commercial solid-phase peptide synthesis. Nevertheless, Boc chemistry is still occasionally used for the synthesis of complex or base-sensitive peptides.
Independently of the Nα-protecting group used in the synthesis, the solid-phase approach has retained its original principle and steps. The process proceeds by coupling the protected amino acid residues with solid-phase, peptide extension can proceed by a series of deprotection and coupling cycles. These reactions employ the use of soluble reagents in excess that can be removed by washing and filtration without manipulative losses.
Due to the fully optimized and inexpensive production of peptides, these biomolecules have found countless uses for a wide range of applications:
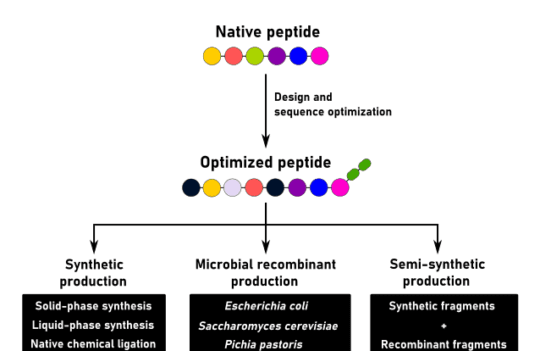
Solid-phase peptide synthesis has enhanced the discovery of new therapeutic peptides and has allowed the use of these molecules for diagnostics and drug discovery.
Despite the recent advances in recombinant technologies, this chemical process continues to be the preferred way to synthesize high-purity peptide formulations at large-scale and commercial levels.
Join our email list to receive exclusive content featuring the most interesting industry and research news, biologics development tips pieced together by experts, res, company news, and exclusive limited-offers. Join a community of 80,000 subscribers and save up to 30% on your first order.